Genotyping¶
Defining Locus types and attributes¶
A Locus type is a category of locus or genetic markers with common descriptors. It is one of the descriptors of an Locus. For instance, a locus type can be SNP, SSR, RFLP etc.
Locus type can be managed in the menu Admin > Genotyping > Locus type.
It is defined by the fields described in the table below. Fields in bold are required while italic ones are optional when submitting the form.

Locus administration menu¶
Locus types can be edited from the table below the form.
Field name |
Description |
|---|---|
Name |
The name of this locus type (for instance : SNP, SSR, Genomic sequence) |
Description |
A text describing this locus type |
Locus are described regarding a set of fixed attributes (see below), common to all locus types. Thaliadb makes possible to add specific attributes to each locus type. Attributes can be managed in the menu ‘Attributes’ in the left navigation toolbar. An attribute is defined by the fields described in the table below. Fields in bold are required while italic ones are optional when submitting the form. Attributes can be edited from the table below the form.
Field name |
Description |
|---|---|
Name |
The name of this attribute |
Type |
A type defining the nature of the information to choose between : Short text, Long text, Number, URL The type will have consequences on the look of the widget in Locus form |
Description |
A short description for this attribute |
As soon as your attributes are defined you can add them as additional descriptors in locus type. Adding attributes to a locus type will enrich the fields available when you create a new locus of this type. To do so, go back to the Locus type table and edit one of them. A new tab ‘Attributes’ will appear in the edition form.
In this interface, you can add attributes to the current locus type (1). By default, this attribute will be added at the last position. You can change this order by drag’n drop, as soon as you’re fine with your attributes order you can click on the ‘Save order’ button (2). This order that you defined will be respected in the Locus form and when you will export locus data from thaliadb.

Locus type attributes management works as Accession type attributes management¶
Locus positions¶
Locus can be positioned regarding a genome version. The position is filled when the locus is recorded in Thaliadb. New positions can be added to a newer genome version for a set of locus.
This can be done in the menu Admin > Genotyping > Locus Position by selection a genome version and submitting a file with the following fields :
Field name |
Description |
|---|---|
Name |
The name of this Locus |
Chromosome |
If position is known for this locus, the chromosome on which it is positioned |
Position |
The position of this locus if it is known |
Locus management¶
A Locus is a fixed position on a chromosome. This position can be known or not and can be different from a genome version to another.
Locus can be managed in the menu Admin > Genotyping > Locus type. In the left navigation toolbar, a menu is available for each locus type that have been defined in the tool.
It is defined at least by the fields described in the table below (additional fields can be defined as explained in the previous section). Fields in bold are required while italic ones are optional when submitting the form. Locus can be edited from the table below the form.
Field name |
Description |
|---|---|
Name |
The name of this Locus |
Comments |
A description for this Locus |
Genome version |
If position is known for this locus, the genome version on which it is positioned |
Chromosome |
If position is known for this locus, the chromosome on which it is positioned |
Position |
The position of this locus if it is known |
Referential¶
A Referential is a reading of a genotyping experiment regarding a defined context.
referential can be managed in the menu Admin > Genotyping > Referential.
It is defined by the fields described in the table below. Fields in bold are required while italic ones are optional when submitting the form. Referential can be edited from the table below the form.
Field name |
Description |
|---|---|
Name |
The name of this Referential |
Person |
The reference person for this referential |
Date |
The creation date of this referential |
Comments |
Description of this referential |
Linked files |
A set of related documents for this referential |
Projects |
Projects in which the referential is shared |
Experiment¶
An Experiment is the molecular characterization of a panel of Accessions regarding a set of markers.
Experiments can be managed in the menu Admin > Genotyping > Experiment.
It is defined by the fields described in the table below. Fields in bold are required while italic ones are optional when submitting the form. Experiment can be edited from the table below the form.
Field name |
Description |
|---|---|
Name |
The name of this Experiment |
Institution |
The institution that lead this experiment |
Person |
The person that lead this experiment |
Date |
The creation date of this experiment |
Comments |
Description of this experiment |
Projects |
Projects in which the experiment is shared |
Genome versions¶
A Genome version is a new annotation or a new sequence of a given genome taxon. It results in new positions for related locus.
Genome versions can be managed in the menu Admin > Genotyping > Genome version.
It is defined by the fields described in the table below. Fields in bold are required while italic ones are optional when submitting the form. Genome version can be edited from the table below the form.
Field name |
Description |
|---|---|
Name |
The name of this Genome version |
Description |
The description of this genome version |
Sample¶
A Sample is a DNA sample of a seed lot. It is used to characterize genetic material in experiments.
Sample can be managed in the menu Admin > Genotyping > Samplen.
It is defined by the fields described in the table below. Fields in bold are required while italic ones are optional when submitting the form. Sample can be edited from the table below the form.
Field name |
Description |
|---|---|
Name |
The name of this Sample |
Seed lot |
The origin seed lot of this sample |
Description |
The description of this sample |
Is bulk |
Denote a bulk sample |
Is obsolete |
Denote this sample is obsolete |
Code labo |
The code used in the lab for internal identification |
Tube name |
The name used to indentify the sample at the lab |
Projects |
Projects in which the sample is shared |
GenotypingID¶
A GenotypingID is the characterization of a sample in one <experiment.
GenotypingID can be managed in the menu Admin > Genotyping > GenotypingID.
It is defined by the fields described in the table below. Fields in bold are required while italic ones are optional when submitting the form. GenotypingID can be edited from the table below the form.
Field name |
Description |
|---|---|
Name |
The name of this genotypingID |
Sample |
The origin sample of this genotypingID |
Experiment |
The <experiment of this genotypingID |
Sentrixbarcode a |
?? |
Sentrixposition a |
?? |
Funding |
Source funding for this genotypingID |
Projects |
Projects in which the genotypingID is shared |
Insert Genotyping data¶
Genotyping data can be uploaded in Thaliadb with files. The form to submit data files can be found in the menu Admin > Genotyping > insert data.
Multiple formats are available to submit genotyping data : Flat format, IUPAC matrix, 2 letters matrix, Slash matrix, and VCF. They are detailed in the sections below.
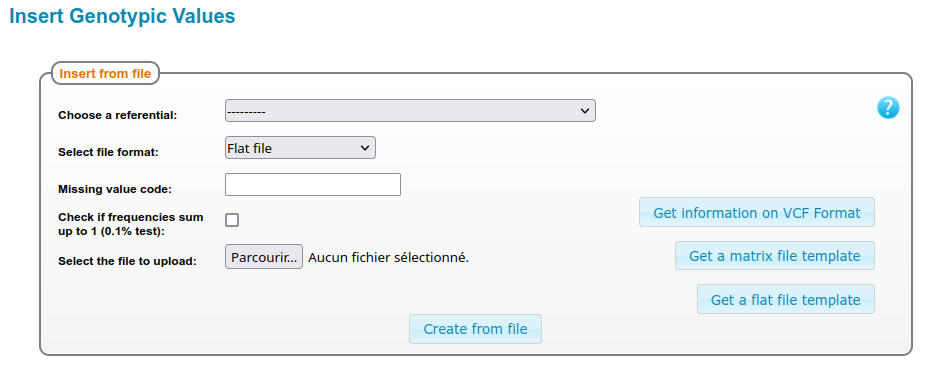
Form to upload Genotyping data¶
Flat format¶
The flat format can be used to describe the genotyping of heterogenous material such as population, Landraces etc. A frequency can be assigned to each allele identified at the locus. The file must contain the following headers :
Field name |
Description |
|---|---|
GenotypingID |
The name of the genotypingID |
Locus |
The name of the locus |
Allele_value |
An identified allele for this genotypinID the locus. |
Allelic_frequency |
A value between 0 and 1 corresponding to the frequency of the allele |
The form to submit genotyping data with flat files provides two options :
missing value : A string meaning an unidentified allele (Any string, any size except the letters ‘A’, ‘C’, ‘T’ and ‘G’).
frequencies test : this option compute a test on all frequencies for one genotypingID at one locus, ensuring that the sum of frequency is 1 +/- 0,001.
Important : missing value will be considered in priority compared with ‘Allele_value’ in the flat file, meaning that you have to take care that your missing value is consistent with the allele values referenced in your file.
IUPAC code matrix¶
The IUPAC code matrix format can be used to describe the genotyping data of material encoded in a one letter. IUPAC code is an established standard. The file is a matrix using standard separators (comma, semi comma, tabulation etc.) of individual vs locus. It is possible to choose if locus are in line or in column.
2 letters code matrix¶
The 2 letters code matrix is more suitable to describe the genotyping data of homogeneous material such as inbred lines. In this file, the genotypes are encoded with 2 letters such as AA, CC, TT, GG if the individual is homozygous for this locus. Heterozygous can also be encoded with 2 letters (for instance AT, CT, GA etc.). Insertion and deletion are encoded with the symbols ‘+’ and ‘-‘.
The form to submit genotyping data with 2 letters files provides two options :
missing value : A string meaning an unidentified allele (Any string, any size except the letters ‘A’, ‘C’, ‘T’, ‘G’, ‘+’ and ‘-‘).
matrix orientation : this option makes possible to choose if locus are in line or in column.
Slash matrix¶
The “Slash” matrix is also suitable to describe the genotyping of homogeneous material such as inbred lines. In this file, the genotypes are encoded with 2 strings separated by a slash. This format offers more flexibility to encode alleles values regarding the 2 letters matrix format.
The form to submit genotyping data with a slash file format provides two options :
missing value : A string meaning an unidentified allele (Any string, any size except the letters ‘A’, ‘C’, ‘T’, ‘G’).
matrix orientation : this option makes possible to choose if locus are in line or in column.
Important : missing value will be considered in priority compared with allele values detected in the cells of the matrix That means you have to take care that your missing value is consistent with the allele values referenced in your file.
VCF format¶
The VCF option can be selected to submit genotyping data with this established standard. No option is available here as Thaliadb follow the format description. The VCF file will be stored in Thaliadb “as it is” and also converted in a matrix format.